HSP90: Regulation
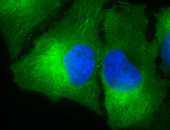
 IHC staining of inflammatory cells and epithelia mucosa in mouse colon tissues, using Anti-Hsp90 (clone: AC-16)
IHC staining of inflammatory cells and epithelia mucosa in mouse colon tissues, using Anti-Hsp90 (clone: AC-16)
When cells are subjected to environmental stress, they respond by enhancing expression of HSPs. The rapid induction of HSP in response to environmental stress is based on a variety of genetic and biochemical processes referred to as the heat shock response (HSR) 13. HSR is regulated mainly at the transcription level by heat shock factors (HSF). HSFs are transcription factors that bind specific cis-acting sequences up-stream of the heat shock gene promoters called heat shock elements (HSE) 203. Among them, HSF-1 is considered as being the key transcription factor of stress-inducible HSPs 204,205. Like any other transcription factors, HSFs are modular proteins composed of several functional domains, including a DNA-binding domain (DBD) and two oligomerization domains (HR-A/B) at the N-terminus, a third oligomerization motif (HR-C) and an activation domain at the C-terminus, and one or more regulatory domains intermingled to these. DBD consists of helix-turn-helix motifs which form compact globular structures that regulate DNA-binding and target gene recognition 206. In contrast, the hydrophobic heptad repeat (HR)-A/B domains mediate both, spontaneous and inducible oligomerization of HSF-1 by forming a coiled coil conformation typical of leucine zipper-containing proteins 207. Under non-stress conditions, HSF-1 exists as an inactive monomer in the cytoplasm due to intramolecular bonds between HR-C and HR-A/B domains and its association with Hsp70 and Hsp90 204. Monomeric HSF-1 is already a phosphoprotein and interacts weakly with Hsp90 and, in response to stress, Hsp90 dissociates from the complex leading to HSF-1 trimerization 89,208. HSF-1 homotrimers bind to HSEs of any HSP and trigger instantaneous and massive transcription of these HSP genes after hyperphosphorylation, a prerequisite for its transcriptional activity 204,207,209. HSF-1 activation is negatively regulated by Hsp90 and Hsp70 via an autoregulatory loop 210. Trimeric HSF-1 is kept inactive when its regulatory domain is bound by a multi-chaperone complex of Hsp90, co-chaperone p23 (PTGES3) and immunophilin FKBP52 88,89,209,211. Activated HSF-1 trimers also interact with Hsp70 and the co-chaperone Hsp40 (DnaJ), leading to an inhibition of its transactivation capacity 212,213,214. HSF-1 transcriptional activity is attenuated not only by the negative feedback loop from HSPs, which represses the transactivation of DNA-bound HSF-1, but also by inhibition of DNA binding through acetylation of Lys80 in the DNA-binding domain of HSF-1. The deacetylase sirtuin-1 regulates the attenuation phase of the HSR by mediating HSF-1 deacetylation 215. HSF-1 activation is also accompanied by sumoylation 216.
Apart from its transcriptional regulation, Hsp90 protein levels have also been found as being regulated at the post-transcriptional level. Gene expression studies in cancer revealed an interruption between transcription and translation of different HSP90s. Tang and collaborators demonstrated that HSP90 mRNA expression and protein expression varied considerably as HSP90 protein levels increased after heat stress while HSP90 mRNA levels remained almost unaffected 217. In this context, the discovery of micro-RNA (miRNA) identified this RNA subtype as a crucial player in regulating translation of many genes. miRNAs are a class of small non-coding RNAs that negatively regulate gene expression by binding to target mRNAs. Global alterations in miRNAs can be observed in a number of disease states including cancer 218,219,220. However, little information is available on the role of miRNAs in the regulation of the HSP90 expression. The group of Biao Cheng identified Grp94 (Hsp90B1) as being a direct target of miR-223 in osteosarcoma acting as a tumour suppressor via the PI3K/Akt/mTOR pathway 221. A recent study convincingly demonstrated that hyperthermia-induced up-regulation of Hsp90 was suppressed by miR-27a in human oral squamous cell carcinoma cells 222. Hsp90β has been found as being an indirect target of miR-499 because miR-499 modified the phosphorylation state of Hsp90β in experimental cardiomyopathy 223. However, future approaches analyzing the regulatory potential of miRNAs in HSP90 expression will shed light on the post-transcriptional regulation of HSP90s.
Hsp90 may be also expressed irrespectively of HSF-1 transcriptional activity. It has been shown previously that interleukin-6 (IL-6) increases the levels of Hsp90 and activates the Hsp90β (HSP90AB1) promoter via the IL-6-activated transcription factors NF-IL6 (C/EBPβ) and signal transducer and activator of transcription 3 (STAT-3). Binding of IL-6 to its receptor stimulates distinct signalling pathways culminating in the activation of NF-IL6 (C/EBPβ) and STAT-3 224. Both factors activate the Hsp90β (HSP90AB1) promoter and synergistically affect its transcription resulting in its observed activation by IL-6. Interestingly, activation of the HSP promoters by these factors is mediated via NF-IL6 and STAT-like binding sites located close to the HSEs 225. Additionally, the two transcription factors interact differently with HSF-1 and heat shock stress 225. Inasmuch as STAT-3 activation reduced the stimulatory effect of HSF-1 or heat stress, activation of NF-IL6 enhanced it indicating that a specific stimulus such as IL-6 can up-regulate HSP expression regardless of stress in an HSF-1-independent manner and that others can functionally interact with HSF-1 in HSP gene regulation. A further candidate for Hsp90 synthesis in the absence of stress comprises the transcription factor STAT-1, which has been reported to mediate the expression of Hsp90 in human fibroblasts exposed to IFNγ 226. In addition, over-expression of STAT-1 was found to enhance the activity of the Hsp90β (HSP90AB1) promoter. The effect of IFNγ/STAT-1 was mediated via a short region of the Hsp90β promoter which also mediates the effects of NF-IL6 and STAT-3 and binds STAT-1. This region further contains a binding site for the stress-activated transcription factor HSF-1. Notably, STAT-1 and HSF-1 were shown to have an additive effect in activating the Hsp90β (HSP90AB1) promoter and to directly interact via a protein-protein interaction. Together, these studies have identified a composite response element within the HSP90 proximal promoter region that integrates the HSF-mediated heat shock response with IL-6 and IFN-γ signalling to mediate the differential regulation of HSPs.
It has been shown previously that several inflammatory mediators and signalling molecules such as NF-κB and TNF are strictly bound to chaperone / HSP gene expression and protein functions. In this context, the NF-κB subunit p65/RelA functions as a transcription factor for numerous HSPs including Hsp90 227,228,229,230 that in turn may have anti-apoptotic functions 8,230,231. The study by Ammirante and colleagues convincingly demonstrated that NF-κB binds to one of the two putative consensus sequences present in the Hsp90α (HSP90AA1) promoter (not in the Hsp90β/HSP90AB1 promotor) suggesting the existence of a regulatory loop between NF-κB and HSPs able to affect cell survival and stress responses 230.
Hsp90 activity is regulated by several post-translational modifications including phosphorylation, acetylation, oxidation, and S-nitrosylation 232 as well as methylation, O-glycosylation, ubiquitination and sumoylation 232,233,234.
Phosphorylation was the firstly identified Hsp90 post-translational modification in mammalian cells 235,236. Hsp90 is specifically phosphorylated at certain serine, threonine and tyrosine residues affecting function and interaction of client proteins with Hsp90 237,238. Early work showed that the serine/threonine phosphatase inhibitor okadaic acid promoted Hsp90 hyperphosphorylation accompanied by reduced association with its client kinase pp60v-src implying a link between Hsp90 phosphorylation and chaperoning of its client proteins 239. The steroid hormone receptor-associated phosphatase PP5/Ppt-1 was found to dephosphorylate Hsp90 as well as the receptor itself 240,241. In contrast, diminished phosphorylation of Hsp90 as can be found in some types of leukemias was shown to encourage the association of Hsp90 with apoptosis protease-activating factor 1 (Apaf-1) leading to inhibition of cytochrome c-induced apoptosome activation that may contribute to chemoresistance in leukemias 242. Hsp90 is also a substrate for DNA-dependent protein kinase, Akt, B-Raf, casein kinase II, and PKA 243,244,245,246,247. The latter has been identified to induce phosphorylation of Thr90 in human Hsp90α thereby increasing the association of Hsp90α with the client protein eNOS 243.
However, the influence of Hsp90 phosphorylation on its activity is far from being completely understood. Previous work identified the conserved Tyr38 in human Hsp90α and Tyr24 in yeast Hsp90, respectively, as the primary target of the tyrosine kinase Wee-1Swe-1 248,249. The same group found out that blockage of the Wee1Swe-1-mediated Hsp90α phosphorylation inhibited Hsp90 interactions with certain co-chaperones (Aha-1) and client proteins (Ste-11, Mpk-1/Slt-2) 248. In addition, Mollapour and colleagues reported that casein kinase II phosphorylates the conserved Thr22 in α-helix 1 of the yeast Hsp90 N-domain thereby modulating its interaction with the co-chaperones Cdc-37 and Aha-1, respectively 237.
Evidence for an acetylation of Hsp90 has emerged after the discovery of the histone deacetylase 6 (HDAC6) as being an interaction partner of Hsp90, a key component of the GR chaperone complex 250,251,252,253,254,255. HDAC6 has previously been shown to regulate GR chaperoning via reversible acetylation of Hsp90 250,253,254,256. In this context, hyperacetylation of Hsp90 following HDAC6 depletion has been shown to alter the assembly of the GR/chaperone complex and impair downstream cellular responses to glucocorticoid hormones as Hsp90 hyperacetylation promotes dissociation of p23 from the GR/Hsp90 complex 253, 254. Consequently, GR looses its capacity for steroid binding or translocates into the nucleus, respectively 253,254. Several other HDACs, such as HDAC1 and HDAC10, can also deacetylate Hsp90 257,258,259. Hsp90 is known to be hyperacetylated at eleven lysyl residues culminating in blockage of ATP-binding and its interaction with several client proteins. Treatment of human breast cancer cells with the HDAC inhibitor LBH589 has been shown to induce hyperacetylation of Hsp90, thereby inhibiting the association of Hsp90 with DNA methyltransferase 1 (Dnmt-1) and promoting its ubiquitination 260. Moreover, knockdown of HDAC1 was associated with Hsp90 acetylation in conjunction with reduction of Dnmt-1 protein expression 260. Dnmt-1 represents a highly active epigenetic DNA modifier which plays an important role in silencing tumour suppressor genes via DNA methylation.
S-Nitrosylation and oxidation are further post-translational modifications of Hsp90 occurring preferentially at Cys597 located to the CTD and affecting conformation and function of Hsp90 261,262,263. S-nitrosylation affects the dimerization dynamics and activity of Hsp90 by abrogating its ATPase activity and affinity to client proteins 261. Similar effects are observed in the presence of reactive oxygen species (ROS). It has been shown previously that glutathione depletion abrogated mineralocorticoid receptor-mediated biological responses 264,265. Notwithstanding the functionality of the SHR is blocked under oxidative conditions, it can be restored under reducing conditions 266,267. Steroid-induced ROS formation has been found to inhibit the activity of the Hsp90-Hsp70 chaperone complex, at least in part, by a direct thiol oxidation of Hsp90 and Hsp70, which in turn leads to the destabilization and depletion of Hsp90 client proteins thereby causing cell cycle arrest and apoptosis 268. These data clearly indicate that the effects of the SHRs are dependent on Hsp90, and oxidation of HSPs attenuates cell proliferation.
Human Hsp90α is methylated by the lysine methyltransferase Smyd-2 on Lys209 and Lys615 (Lys616 in mouse), which is inhibited by Hop, and Lys615 is demethylated by the lysine amine oxidase Lsd-1 suggesting that alternative mechanisms control Hsp90 methylation by Smyd-2 269. Hsp90β was also found to be methylated by Smyd-2 on Lys616, equivalent to Lys607 in humans 270. In muscle, Hsp90 methylation was reported to contribute to the formation of a protein complex containing Smyd-2, Hsp90, and the sarcomeric protein titin. Deficiency in Smyd-2 resulted in the loss of Hsp90 methylation, impaired titin stability, and altered muscle function. Collectively, these data reveal a cytoplasmic protein network that employs lysine methylation for the maintenance and function of skeletal muscle 270.
Several tyrosine residues can be nitrated of which Tyr33 or Tyr56 in Hsp90β are sufficient to inhibit ATPase activity and to display a dominant toxic gain of function. Nitrated Hsp90 binds and activates the ATP-gated ion channel P2X7 thereby eliciting apoptosis, for example in motoneurons 271. The toxic form of nitrated Hsp90 is present in vivo in pathological conditions such as amyotrophic lateral sclerosis (ALS) and spinal cord injury. The data presented by Franco et al. provide conclusive evidence that selective and limited tyrosine nitration plays a causal role in the induction of cell death 271.
Post-translational modifications of Hsp90 also include O-glycosylation as well as sumoylation. In murine Hsp90β, two novel O-glycosylation sites could be identified of which one corresponds to a previously described phosphorylation site 234. The group of Len Neckers currently showed that asymmetric sumoylation of the conserved Lys178 in the N-domain of yeast Hsp82 and the conserved Lys191 of human Hsp90α promotes both, interaction with the co-chaperone Aha-1 and binding of Hsp90 inhibitors 233. Sumoylated Hsp90 has higher affinity for inhibitors, which increase sumoylation, compete with Aha-1, and displace ATP prior to lid closure (Mollapour et al., 2014). Importantly, cellular transformation is accompanied by elevated steady-state N-domain sumoylation, and increased Hsp90 sumoylation sensitizes yeast and mammalian cells to Hsp90 inhibitors, providing a mechanism to explain the sensitivity of cancer cells to these drugs. Unlike in yeast, Lys191 of human Hsp90α is also subject to ubiquitination and acetylation 233. Moreover, the ubiquitin ligase Hectd-1 has been found to polyubiquitinate Hsp90α via Lys63 linkages thereby preventing its secretion 272. The same study demonstrated that Hectd1-dependent polyubiquitination of Hsp90 affects its intracellular location and negatively regulates its secretion, as in the absence of Hectd-1 ubiquitin ligase activity, extracellular Hsp90 levels are increased. Furthermore, the disorganized mesoderm-derived cranial mesenchyme (CM) in HECDT1 mutant embryos, a potential underlying cause of exencephaly, is most likely related – at least in part – to the abnormal behavior of CM cells stimulated by the excess Hsp90 secreted from mutant cells 272. In yeast cells, Swe-1Wee-1-mediated phosphorylation of Hsp90 signals Hsp90 polyubiquitination and subsequent degradation by cytoplasmic proteasomes 248. The ubiquitin ligase C-terminus of Hsc70 interacting protein (CHIP) has been shown in vitro to ubiquitinate human Hsp90β at 13 lysine residues thereby forming polyubiquitin chains with Hsp70 linked via Lys6, Lys11, Lys48, and Lys6 273. These data shed light on the mode of ubiquitination of Hsp70 and Hsp90 by CHIP, which ultimately leads to their degradation in the proteasome.