HSP90: Inhibitors
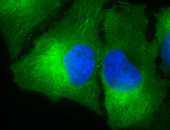
 IF detection of Hsp90 in cancerous mouse colon tissue, using Anti-Hsp90 (clone: H9010)
IF detection of Hsp90 in cancerous mouse colon tissue, using Anti-Hsp90 (clone: H9010)
Many oncogenic proteins essential for the transformation of cells are client proteins of Hsp90. Therefore, targeting Hsp90 with chemical inhibitors would degrade these oncogenic proteins and thus function as anti-cancer agents. Hsp90 inhibitors deplete Hsp90 client proteins in cancer cells without having any effect on their cellular counterparts in non-transformed cells 152,154. The molecular basis for the selective anti-tumoural activity of HSP90 inhibitors appears to relate to the conformation of the Hsp90-inhibitor complex as Hsp90 isolated from tumour cells has a 20 to 200 times higher binding affinity for the inhibitor than Hsp90 from non-transformed cells 154. HSP90 inhibitors have the potential for use in single-agent or combinatorial therapies in order to supplement the hitherto existing conventional chemotherapeutical approaches and molecularly targeted drugs. All molecules derived from the benzoquinone ansamycin geldanamycin are designated as “first-generation” Hsp90 inhibitors while all other fully or semi-synthetic drugs are referred to as “second-generation” inhibitors.
Natural inhibitors of Hsp90 and their derivatives
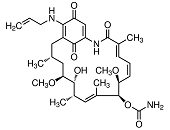
In most instances, natural inhibitors of Hsp90 interact with high affinity with the ATP-binding site of Hsp90 thereby blocking the cycling between ADP- and ATP-bound conformations and impairing its chaperoning capacity 437. The benzoquinone ansamycin geldanamycin was about the first Hsp90 inhibitor with promising anti-tumour properties in preclinical settings. Unfortunately, geldanamycin showed an unfavourable solubility and toxicity leading to the suspension of Phase I clinical trials 438 and the development of less toxic derivatives such as 17-allylamino-17-desmethoxygeldanamycin (17-AAG, tanespimycin) 439. These agents were found to destabilize and degrade virtually all Hsp90 co-chaperones in vivo and showed promising anti-cancer properties in preclinical model systems 334. In mantle cell lymphoma cell lines, 17-AAG induces cell cycle arrest and apoptosis by activating caspase 9 and depleting cyclin D1, Akt, and Bid 440. 17-AAG, the first-in-class Hsp90 inhibitor has completed phase I trials and entered phase II trials 441. However, 17-AAG has a poor solubility, lacks oral bioavailability, and shows moderate clinical anti-tumour activity only 389. 17-DMAG (alvespimycin) is an N,N-dimethylethylamino analog of 17-AAG with improved solubility that entered phase I clinical trials where a tolerable toxicity was noted 442,443. In breast cancer, 17-DMAG was reported to mediate its anti-tumour effect through down-regulation of receptor interacting protein 1 (Rip-1) thereby sensitizing breast cancer cells to TRAIL-induced apoptosis 444. Models of murine medulloblastoma that retain and lack p53 function, respectively, displayed a dependence on functional p53 to engage 17-DMAG-induced apoptosis 445. In multiple myeloma, 17-DMAG attenuates STAT-3 and phospho-ERK levels that critically contribute to tumour cell survival in an IL-6R/STAT-3 and MAPK-dependent matter, respectively 446.
The novel potent and selective HSP90 inhibitor IPI-504 (retaspimycin hydrochloride) is an analog of 17-AAG with improved water solubility suitable for parental administration. Preclinical studies and early clinical trials have confirmed the efficacy of IPI-504 in NSCLC both as monotherapy and in combination with docetaxel 447. A phase I trial of IPI-504 in patients with gastrointestinal stromal tumours (GIST) and soft-tissue sarcomas (STS) revealed stable disease (SD) in 70% of patients with GIST and 59% of patients with STS 448. There was one confirmed partial response (PR) in a patient with GIST and one PR in a patient with liposarcoma. Metabolic partial responses occurred in 38% of patients with GIST. In breast cancer, a multicenter phase I trial induced 62% of stable disease after treatment with IPI-504 in combination with trastuzumab in patients with locally advanced or metastatic Her-2-positive breast cancer 449. Further phase I/II trials are currently underway to evaluate the dosing schedules and activity of IPI-504 in breast cancer 450. Moreover, a phase I dose escalation of IPI-504 revealed stable disease in 22% of patients with relapsed or relapsed and refractory multiple myeloma 451. Given the in vitro activity in diffuse large B-cell lymphoma, mantle cell lymphoma, melanoma, leukemia and pancreatic cancer, current and future trials are of clinical interest. Notably, IPI-504 is the only first-generation Hsp90 inhibitor still under active development. Recently, a related compound, the orally bioavailable IPI-493 [17-amino-17-demethoxygeldanamycin (17-AG)] has shown promising results in preclinical models 452. A phase I dose escalation study of IPI-493 in patients with advanced malignancies has been terminated as drug exposure of IPI-504 has been found as being superior to IPI-493.
Combinatorial administrations of geldanamycin derivatives and other chemotherapeutics show promising results in several preclinical approaches. The receptor tyrosine kinase Bcr-Abl, which is over-expressed in chronic myeloid leukemia (CML), is a client protein of Hsp90 and can be inhibited by imatinib mesylate (STI-571) rendering cancer cells resistant to apoptosis 453. A combination of imatinib and 17-AAG has been reported to act synergistically in down-regulating Bcr-Abl protein levels and inducing apoptosis in Bcr-Abl-overexpressing CML cells 454. This combinatorial treatment has been evaluated in a phase I trial in CML patients with no results reported up to date. Geldanamycin derivatives were also used in combination with the proteasome inhibitor bortezomib 455, the apoptosis inductor arsenic trioxide 456, and the DNA-damaging agent doxorubicin 457. Such combinatorial treatments have emerged as promising therapeutic approaches in distinct malignancies.
The coumarin-related antibiotic novobiocin represents a further natural Hsp90 inhibitor interacting with the CTD of the chaperone thereby disrupting Hsp90 dimerization 458,459,460,461. Novobiocin has been introduced into clinical use against Staphylococcus aureus infections including multiresistant MRSA strains. Novobiocin is also active against Borrelia burgdorferi, the causative agent of Lyme disease 462. Novobiocin has been shown to destabilize several Hsp90 client proteins including Bcr-Abl, Her-2, mutant p53, and Raf-1 461. In Bcr-Abl-positive human leukemia cells, disruption of the Hsp90/Bcr-Abl complex by novobiocin induces cytosolic accumulation of cytochrome c and activation of caspase-9 and caspase-3, followed by apoptosis induction 463. Novel novobiocin derivatives such as KU135 and F-4 exert potent anti-cancer effects in vitro and in vivo 464,465,466,467.
The naturally occurring resorcylic acid lactone radicicol has originally been shown to target Hsp90 by binding to its N-terminal NTD 105, 468. In addition, radicicol depletes Hsp90 client signalling molecules in cells, and thus inhibits signal transduction pathways 469. Although radicicol itself displays strong anti-viral and anti-tumour properties in vitro 470,471, it is rapidly converted into metabolites that exhibit little or no affinity for Hsp90 in vivo 472. Consequently, radicicol failed to elicit potent anti-tumour activities against human tumour xenograft models 469. Unlike radicicol itself, its oxime derivatives including KF58333 showed potent anti-tumour activities against human tumour xenograft models. Hsp90 client proteins such as Bcr-Abl and Raf-1 were depleted and apoptosis was induced in the tumour specimen treated with KF58333 473. Apart from these resorcinol-based pyrazole inhibitors, isoxazole-based inhibitors including NVP-AUY922 (VER-52296) have been reported to inhibit several HSP90s with different affinities thereby eliciting potent anti-tumour actions in tumour xenografts 474,475. A phase I trial of NVP-AUY922 in patients with advanced solid tumours demonstrated acceptable tolerability with prolonged disease stabilization in some patients 474,475. Meanwhile, phase II single-agent and combination studies have been initiated in patients with Her-2-positive breast, gastric and non-small cell lung cancers.
(−)-Epigallocatechin gallate (EGCG) is the primary flavonoid component of green tea and has been widely studied due to its diversity of biological properties including anti-inflammatory, anti-oxidative, anti-cancer, anti-infective and neuroprotective activity 476,477,478. EGCG was characterized previously as an Hsp90 inhibitor which binds at or near to a C-terminal ATP-binding site of Hsp90 thereby preventing dimerization 479. In human ovarian carcinoma SKOV3 cells, EGCG modified the association of Hsp90 with several co-chaperones and decreased levels of several cancer-related Hsp90 client proteins, such as Erb-B2, Raf-1 and phospho-Akt 479. Unfortunately, EGCG has poor drug-like properties because of its chemical and metabolic instability responsible, amongst others, for its low bioavailability. Recent approaches to develop novel more drug-like derivates of EGCG led to the identification of selected compounds that were more potent than EGCG in an Hsp90 functional assay 480.
Synthetic inhibitors of Hsp90
Synthetic molecules designed to evade the toxic side effects attributed to the benzoquinone group on geldanamycin-based molecules have been developed and show promising preclinical anti-tumour activity 481,482. XL888 represents a novel small molecule inhibitor of Hsp90 that has been reported to overcome resistance to B-Raf inhibitors (such as vemurafenib and debrafenib) in preclinical models by blocking the expression and/or functional activity of Hsp90 client proteins crucial for growth and cell cycle re-entry (e.g. Akt, A-Raf, C-Raf, mutated N-Ras, , IGF-1R, cyclin D1, PDGFRβ, the MAP kinase family member COT) as well as by inducing the pro-apoptotic Hsp90 client protein, Bim (Bcl-2 interacting mediator of cell death) and by down-regulating Mcl-1 483.
Several other small molecule Hsp90 inhibitors have been reported in clinical settings including AUY922, AT13387, STA9090, and MPC3100. In a phase I dose escalation study of the isoxazole-based Hsp90 inhibitor AUY922, disease stabilization was noted in 16% and partial metabolic response in 9% of advanced solid tumour patients 484. Based on these findings, phase II single-agent and combination studies have been initiated in patients with Her-2-positive breast, gastric and non-small cell lung cancers. Preliminary data from phase II clinical trials of AUY922 have shown responses as a single agent in cancers driven by mutated or over-expressed EGFR in NSCLC 485 or Her-2 in breast cancer 486. Responses have also been described in a Phase IB/II study of AUY922 in combination with trastuzumab in Her-2-positive advanced breast cancer 487 as well as in a phase II trial of AUY922 and the EGFR inhibitor erlotinib in patients with EGFR-mutant lung cancer and acquired resistance to EGFR tyrosine kinase inhibitors 488. AT13387 is a highly potent Hsp90 inhibitor derived from Astex fragment chemistry platform, Pyramid. AT13387 suppresses client proteins for greater than 7 days in tumour cells, making it the longest acting agent reported to date. A phase I study in patients with refractory solid tumours revealed an astonishing toxicity of reversible visual changes including blurred vision, flashes and delayed light/dark accommodation 489. In this study, single-agent AT13387 elicits a dose proportional pharmacodyamic inhibition of cyclin-dependent kinase 4 (CDK4), Raf-1 and phospho-Akt in peripheral blood mononuclear cells (PBMCs). However, the best response in this pre-treated population has been stable disease that persisted for at least six months in two patients (follicular cell carcinoma of the thyroid and metastatic uveal melanoma) 489.
In another phase I study, administration of the second-generation Hsp90 inhibitor ganetespib (STA-9090) has been reported as being broadly accepted in patients with solid tumours with dose-limiting toxicities of amylase elevation, fatigue, nausea, anemia, diarrhea, vomiting, anorexia, and headache. Preliminary signs of clinical activity have been observed in several patients with different tumour types including NSCLC, renal cell carcinoma, melanoma, rectal carcinoma and GIST 490,491,492. In tumour samples from patients with rectal cancer, significant down-regulation of PDGFA, FGF2, ANG1, ANG2, TGFB1, VEGF, STAT3 and HIF1A mRNA was observed after ganetespib treatment 493 . A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced NSCLC induced partial response in 4% of the patients with a manageable side effect profile 391. MPC-3100 is a novel purine-based orally bioavailable small-molecule Hsp90 inhibitor 494. A phase I trial of MPC-3100 yielded a dose-limiting toxicity of supraventricular tachycardia and respiratory failure in a single patient leading to further dose expansion to determine the safe dose 495.
Another group of synthetic Hsp90 inhibitors comprises purine-based compounds. Such inhibitors compete with ADP/ATP for the ATP-binding site inside of Hsp90 thereby inhibiting chaperone functions. PU-H71 is a water-soluble member of the purine class of Hsp90 inhibitors which demonstrated putative efficacy in triple negative breast cancer models 496. As demonstrated by Caldas-Lopes et al., PU-H71 down-regulated components of the Ras/MAPK pathway thereby mediating anti-proliferation, degraded Akt and Bcl-xL to induce apoptosis, and inhibited activated NF-κB, Akt, ERK-2, Tyk-2, and PKC to reduce the invasive potential of triple negative breast cancers both, in vitro and in vivo 496. PU-H71 was found to preferentially accumulate in diffuse large B cell lymphomas (DLBCLs) compared to normal tissues and to selectively suppress DLBCLs in vivo in a manner dependent on the Hsp90 client protein Bcl-6, culminating in re-activation of key Bcl-6 target genes and apoptosis. PU-H71 also induced cell death in primary human DLBCL specimens 497. However, clinical trials with PU-H71 are yet to come.
CNF-2024 (BIIB021) is another synthetic orally administrable purine-scaffold Hsp90 inhibitor which binds competitively with geldanamycin to the ATP-binding pocket of Hsp90 and putatively down-regulates Her-2 in vitro and in several human tumour xenograft models in vivo 498. CNF-2024 decreased Hodgkin lymphoma cell viability via NF-κB inhibition and up-regulated ligands for the activating NK cell receptor NKG2D on Hodgkin’s lymphoma cells thereby sensitizing the cells to NK cell-mediated killing 499. It is noteworthy that CNF-2024 exhibited a strong anti-tumour effect outperforming 17-AAG, either as a single agent and or in combination with radiation, thereby improving the efficacy of radiation in head and neck squamous cell carcinoma (HNSCC) models 500. This study renders administration of CNF-2024 alone or in combination with radiation a putative novel therapeutic approach in HNSCC. Interestingly, CNF-2024 was found as being considerably more active than 17-AAG against adrenocortical carcinoma, both in vitro and in vivo, due to the expression of multidrug resistant proteins such as P-gp and Mrp-1 501. The first completed phase I study of this molecule has shown activity in chronic lymphocytic leukemia (CLL), possibly due to depletion of the client protein zeta-associated protein of 70 kDa (Zap-70) 483,502. A more potent, intravenous version of CNF-2024 is currently being tested in a phase I study in advanced solid tumours.
Several other types of Hsp90 inhibitors such as the 2-aminobenzamide derivative SNX-5422 have been developed and successfully tested in multiple myeloma 503 as well as in breast cancer 390 in vitro and in vivo. Despite early promise, results of phase I clinical studies of SNX-5422 in patients with hematologic and solid tumour malagnancies have been disappointing 504, 505. SNX-25a, with a 2-(trans-4-hydroxy-cyclohexylamino) side chain, is a novel 2-aminobenzamide inhibitor of Hsp90 optimized by structure–activity relationship explorations for high Hsp90 affinity 402,506. SNX-25a elicits a much higher efficacy than 17-AAG on growth inhibition of numerous cancer cells 507. The mode of action of anti-tumour activity is obviously associated with the induction of cell cycle arrest, apoptosis and Hsp90 client proteins degradation. This superiority effect of SNX-25a warrants further confirmation in animal models.
Another inhibitor of Hsp90 represents the cell-permeable peptidomimetic shepherdin which targets the ATP-binding pocket of Hsp90 thereby antagonizing the interaction with the Hsp90 client survivin, an anti-apoptotic and mitotic regulator 508. Although shepherdin selectively induces death of tumour cells in vitro, it does not affect colony formation of purified hematopoietic progenitors. These anti-tumour effects of shepherdin could also be demonstrated in mice where shepherdin was shown to inhibit human tumour growth in mice without toxicity 508. An AlphaScreen technology based high-throughput in vitro screen identified several compounds harbouring in common a 7-azapteridine ring system (pyrimido[5,4-e] [1,2,4]triazine-5,7-dione) 509. These compounds act by disrupting the interaction between Hsp90 and its co-chaperone Hop consequently inducing cell death in human breast cancer cell lines through down-regulation of Her-2.
Additional Hsp90 inhibitors are 5′-N-ethylcarboxamideadenosine (NECA), an adenosine analogue, which targets ER-resident Grp94 510, the 3,4-diarylpyrazole CCT-018159 511,512, and its pyrazole amide analogs VER-49009 and VER-50589 482. CCT-018159 was reported to inhibit Hsp90β by depleting the client proteins Erb-B2, Cdk-4, C-Raf, and mutant B-Raf accompanied by an up-regulatoin of Hsp70 in human cancer cell lines 481. Similarly, VER-50589 and VER-49009 caused induction of Hsp70 and Hsp27 alongside depletion of client proteins, including C-Raf, B-Raf, and survivin, and the protein arginine methyltransferase Prmt-5 482.
An interesting variant of the alternative Hsp90 inhibitors is represented by a monoclonal antibody directed against fungal Hsp90. In a randomized blinded multicenter trial of amphotericin B alone vs. amphotericin B in combination with the anti-fungal Hsp90 antibody mycograb, the combinatorial administration produced significant clinical and culture-confirmed improvement in outcome for patients with invasive candidiasis 513.